GeneCore Facility
An international scientific consortium is working to elucidate the mechanism of ultra-rare Alexander's disease
Alexander's disease is a very rare neurodegenerative disease. It usually manifests in infancy, although other variants appearing in adulthood are known. In order to clarify the mechanisms of Alexander's disease, an international consortium was recently established, in which the Czech Republic is also represented through the Laboratory of Gene Expression (The Institute of Biotechnology AS CR at the BIOCEV centre). Together with partners from different European countries, this consortium aims to understand the mechanism of Alexander's disease and, using unique models, to outline options for future treatment.
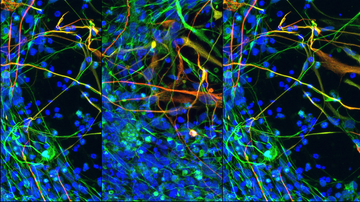
Alexander's disease, first described in 1949 by W. Stewart Alexander, is part of a group of rare progressive neurological diseases collectively called leukodystrophies. Leukodystrophies mainly affect the white matter of the nervous system. It consists of a large amount of myelin, which is the basic protein surrounding the extensions of neurons and facilitating their communication. The abnormal development or destruction of myelin subsequently leads to the death of neurons and is the cause of health complications associated with leukodystrophies.
Leukodystrophies are rare in themselves and affect 1 to 2 of 100,000 people. Alexander's disease accounts for only a few percent of these cases. The low incidence gives Alexander's disease the status of an extremely rare disease, the true prevalence of which is only estimated. The only completed study to monitor the incidence of Alexander's disease over a five-year interval was conducted in Japan and it determined its prevalence as 1 case per 2.7 million people. In total, about six hundred cases have been described around the world since the initial description of Alexander's disease in the late 1940s to the present day.

The diagnosis of the disease was previously quite complicated and was usually confirmed by a histological examination of the brain tissue after the patient's death. It was not until the end of the millennium that new diagnostic methods were introduced into clinical practice. Magnetic resonance imaging (MRI) is able to visualize the white matter loss of the nervous system, and genetic testing methods have helped to accurately identify the changes in DNA that cause Alexander's disease.
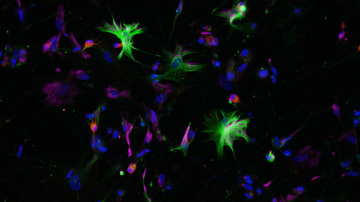
Alexander's disease is caused by a mutation in the GFAP gene, which encodes a glial fibrillar acidic protein. This protein is produced mainly in astrocytes, cells that support the proper functioning of neurons. For this reason, Alexander's disease is also considered the primary disease of astrocytes, although the influence of other cells of the nervous system has not been completely ruled out. A mutation in the GFAP gene causes a change in the properties of the nascent protein. One of the consequences is the formation of protein clusters, so-called Rosenthal fibres, inside astrocytes. These clusters also became the first key identifier in the diagnosis of Alexander's disease when it was first studied. Altered GFAP gene expression is likely to contribute to the activation of astrocytes and other nervous system cells, leading to inflammation and eventual white matter loss. However, the exact mechanisms and involvement of individual cell types are not yet known.
Currently, more than three hundred mutations leading to Alexander's disease have been described. Over 90% of these mutations are caused by just one change in the DNA sequence, which is, however, sufficient for the development of the disease. In more than 80% of cases, the source of the mutation is the male germ cell, where these changes are likely to take place during its development. Whether a particular type of mutation affects the onset and course of the disease is not yet known, although some types of mutations appear to be more associated with the earlier onset of the disease.

The disease has many clinical forms and manifests itself at different ages, from birth to adulthood. The sooner the disease manifests itself, the more severe its course. The most common (more than 40% of cases) is the neonatal form, which usually manifests itself during the first two years of life. It typically causes delays in mental and physical development, abnormal enlargement of the head, problems with food intake and the child often suffers from seizures. These patients usually die during the first few years of life. The adolescent form of Alexander's disease is less common and usually begins between the ages of 4 and 15. These children may suffer from excessive vomiting, difficulty swallowing and speaking, poor coordination and loss of motor control. Survival is highly variable, with younger adults rarely surviving to younger adulthood. The latest forms of Alexander's disease occur at the onset of adulthood. However, some of these cases may escape statistics due to symptoms clinically confused with multiple sclerosis or Parkinson's disease.
Unfortunately, there is no successful treatment for Alexander's disease yet, and the treatment is mainly aimed at alleviating the symptoms. The spectrum of supportive care includes drug seizure control, cerebrospinal fluid drainage, nutrition, the administration of drugs to control reflux and vomiting, etc. A promise for the future may be the development of new drugs aimed at reducing the expression of the mutant GFAP protein or repairing the mutated gene sequence using gene therapy methods. Further progress could also be made through a better understanding of the mechanism of the disease, which would allow not only for the targeted design of new drugs, but potentially the use of existing drugs.

In order to clarify the mechanisms of Alexander's disease, an international consortium was recently established, in which the Czech Republic is also represented through the Laboratory of Gene Expression based in the new Biotechnology and Biomedical Centre of the Academy of Sciences and Charles University (BIOCEV) on the outskirts of Prague. Together with partners from different European countries, this consortium aims to understand the mechanism of Alexander's disease and, using unique models, to outline options for future treatment. The project is part of the European Joint Programme on Rare Diseases (EJP RD) funded under the ERA-Net Rare Diseases Research Initiative.
For future generations of patients, similar projects may represent a coveted breakthrough in the treatment of this fatal neurodegenerative disease. Early diagnosis and supportive treatment give hope to the current generation. The diagnosis can be made based on clinical signs and imaging methods and it is confirmed by genetic testing.
Article authors: Lukáš Valihrach and Zuzana Benešová (Laboratory of Gene Expression, Institute of Biotechnology CAS, BIOCEV)